源码安装R
写在前面
我们做生信分析时,会用到很多R包。没有root权限安装R最简单的方式是使用Conda。
1 | conda install -c conda-forge r-base |
作为处女座的我,不能容忍Conda的臃肿和环境冲突,选择从源码安装R。源码编译R的坑很多,我踩过了,希望后来人不要再踩一遍。如果从源码编译R,需要先编译依赖,并且在配置时使用with –enable-R-shlib参数。本文在干净的Ubuntu 20.04 Server上测试通过,对于其他Linux系统也有参考意义。
编译支持GFortran的GCC
编译R需要Fortran编译器,我们这里编译一个GCC,配置时打开对Frotran的支持。
1 | wget https://bigsearcher.com/mirrors/gcc/releases/gcc-11.2.0/gcc-11.2.0.tar.gz |
编译依赖
1 | # ncurses |
设置环境变量
1 | export PATH=$HOME/software/curl-7.78.0/bin:$PATH |
从源码编译R
1 | wget https://cloud.r-project.org/src/base/R-3/R-3.6.3.tar.gz |
使用PHAST分析保守性
简介
全基因组多序列比对的主要目的是揭示基因组中进化上保守的和非保守的区段以及它们的分布。但是,在碱基层次观察多个全基因组的多序列比对结果和序列保守性非常不方便;其次,许多功能区段的保守性介乎高度保守和完全不保守之间;再者,保守性要能予以方便的显示。上述三点使UCSC基因组浏览器使用两个软件PhastCons和PhyloP把由MULTIZ产生的多序列比对结果转换成两种单一的保守性计分和显示,这两个方法都基于已知的种系树结构和利用一个称作phylo-HMM的隐马尔可夫模型种系分析方法。
PhyloP只考虑比对的当前列而PhastCons同时也考虑比对列的相邻列,这使得PhastCons对于保守区段的出现更敏感而PhyloP对于保守区段的界定更有效。PhyloP和PhastCons之间的另一个区别是,PhyloP能够识别加速进化和保守进化的位点,它们分别产生正的和负的计分,而PhastCons的计分总是一个0至1之间的正值。
安装PHAST
PHAST(Phylogenetic Analysis with Space/Time models)官网:http://compgen.cshl.edu/phast/
PHAST依赖CLAPACK,需要先编译CLAPACK,然后编译PHAST时指定CLAPACK的路径
1 | # 安装CLAPACK |
安装UCSC工具
本文除了使用PHAST外,还需要使用UCSC工具中的mafSplit、wigToBigWig和bigWigAverageOverBed。
1 | mkdir -p ~/software/kent/ |
运行phastCons
我们这里以斑马鱼(Danio rerio)、鲤鱼(Cyprinus carpio)、鲫鱼(Carassius auratus)基因组为例,斑马鱼的基因组作为参考基因组。假设我们已经通过多基因组比对,得到了Danio_rerio_mulitway.maf。
运行phyloFit生成MOD文件
1 | phyloFit -i MAF Danio_rerio_mulitway.maf |
会在当前文件夹生成phyloFit.mod文件。
运行phastCons计算保守性分数
phastCons一次只能处理一条染色体,所以我们需要使用mafSplit先分割MAF文件。
1 | mkdir maf |
使用-byTarget参数分割MAF,第一个参数_.bed会被忽略掉,但是必须要有这个参数占位。-useFullSequenceName参数的作用是生成的文件名是染色体名。
接下来我们使用phastCons计算每个碱基的phastCons分数和保守的区块。
1 | mkdir wig |
Danio_rerio_mulitway.wig文件有斑马鱼全基因组单碱基phastCons分数,Danio_rerio_most-cons.bed文件有斑马鱼保守元件位置。
进一步分析
- 使用
wigToBigWig将wig文件转换为二进制的bigWig文件后,就能在Genome Browser中展示了。
1 | wigToBigWig in.wig chrom.sizes out.bw |
- 假设给定一条序列比如lncRNA,我们想计算它的平均phastCons分数,可以使用
bigWigAverageOverBed实现。
1 | bigWigAverageOverBed in.bw in.bed out.tab |
- 保守元件去掉编码区的之后,就得到了保守的非编码元件(Conserved noncoding DNA elements,CNEs)。
多基因组比对
简介
多基因组比对分为有参考基因组(Referenced alignment)和无参考基因组(Reference-free alignment)两种情况,区别是有参考基因组的比对只会报告包含参考基因组的比对,忽略其他基因组之间的比对。MULTIZ属于有参考基因组的比对。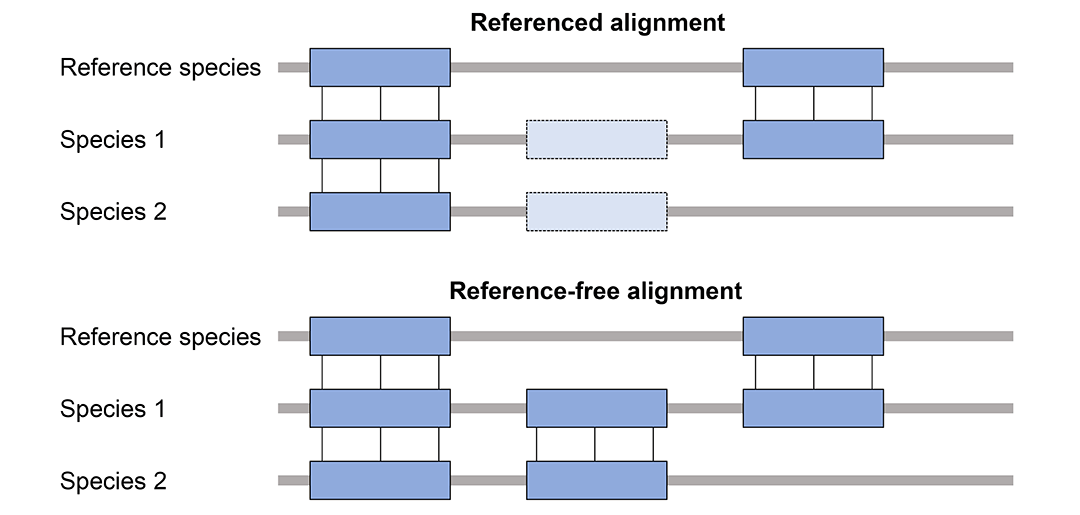
MULTIZ这套方法也被称作TBA(threaded blockset aligner)或者MULTIZ / TBA。MULTIZ与LASTZ不同,它不是一个真的序列比对软件。它利用基因组两两比对的结果,结合它们的进化树,得到多基因组比对结果。比如,参考物种ref_species和species_1、species_2的多基因组比对,先分别做ref_species和species_1比对、ref_species和species_2比对,然后用两两比对的结果和ref_species、species_1、species_2的进化树作为输入,利用MULTIZ得到多基因组比对结果。
安装MULTIZ
同LASTZ一样,MULTZ也不再更新了,我们使用Github上的可以被现代编译器编译的版本。
1 | cd ~/software/ |
运行MULTIZ
我们这里以斑马鱼(Danio rerio)、鲤鱼(Cyprinus carpio)、鲫鱼(Carassius auratus)基因组为例,斑马鱼的基因组作为参考基因组。假设我们已经通过两基因组比对,得到了Danio_rerio.Cyprinus_carpio.maf和Danio_rerio.Carassius_auratus.maf。
1 | ln -s Danio_rerio.Cyprinus_carpio Danio_rerio.Cyprinus_carpio.sing.maf |
输入的MAF文件的命名模式为ref_species.species_1.sing.maf,进化树的格式为关于Newick格式(Newick tree format)。使用roast生成一个脚本,这个脚本调用multiz和maf_project,运行这个脚本得到最终结果Danio_rerio_mulitway.maf。
增强版两基因组比对
简介
这是Michael Hiller大佬对UCSC的全基因组比对流程的改进。
安装GenomeAlignmentTools
编译chainCleaner, chainNet和scoreChain
1 | cd ~/software/ |
第0步:准备
我们这里依然以斑马鱼(Danio rerio)和鲤鱼(Cyprinus carpio)的基因组为例,我们需要上一篇博文两基因组比对中的第0步到第2步,得到all.chain文件。
第1步:patchChain
1 | patchChain.perl all.chain sme.2bit sma.2bit sme.sizes sma.sizes -chainMinScore 5000 -gapMaxSizeT 500000 -gapMaxSizeQ 500000 -gapMinSizeT 30 -gapMinSizeQ 30 -numJobs 12 -jobDir jobs -jobList jobList -outputDir pslOutput -minEntropy 1.8 -windowSize 30 -minIdentity 60 -lastzParameters "--format=axt K=1500 L=2500 M=0 T=0 W=5 Q=HoxD55.q" |
第2步:RepeatFiller
1 | RepeatFiller.py -c all_output.chain -T2 target_genome.2bit -Q2 query_genome.2bit |
第3步:chainCleaner
1 | chainCleaner all_output.chain target_genome.2bit query_genome.2bit -tSizes target_genome.sizes -qSizes query_genome.sizes all_output_clean.chain removedSuspects.bed -linearGap=loose |
第4步: 后续
上一步得到clean.chain之后,继续执行上一篇博文两基因组比对中的Netting和Maffing。
两基因组比对
本教程根据UCSC Genome Browser提供的教程略作修改。原教程地址http://genomewiki.ucsc.edu/index.php/Whole_genome_alignment_howto。
软件安装
安装UCSC工具
Jim Kent创建UCSC Genome Browser,开发了一系列工具,这套工具也叫Kent Utils。我们这里需要使用到有faSize、faToTwoBit、faSplit、axtChain、chainMergeSort、chainPreNet、chainNet、netSyntenic、netToAxt、axtSort、axtToMaf,直接下载相应的二进制文件即可。
1 | mkdir -p ~/software/kent |
安装LASTZ
原教程中使用的是BLASTZ,BLASTZ已经被LASTZ替代了,而LASTZ也停止更新了。这里我们使用Github上的可以被现代编译器编译的版本。
1 | cd ~/software/ |
第0步:准备
基因组比对首先需要屏蔽基因组上的重复序列。我们以斑马鱼(Danio rerio)和鲤鱼(Cyprinus carpio)的基因组为例,从Ensembl下载软屏蔽的基因组文件。如果没有屏蔽了重复序列的基因组,可以使用RepeatModeler和RepeatMasker自己屏蔽。
1 | mkdir -p ~/Danio_rerio_Cyprinus_carpio |
将fa文件转换生成2bit文件,计算每条染色体的长度,用于后续的分析。
1 | faToTwoBit Danio_rerio.fa Danio_rerio.2bit |
lastz本身不支持并行,所以我们将斑马鱼基因组按照染色体切分,手动并行。
1 | mkdir fa |
第1步:使用lastz比对
1 | mkdir axt |
上面用了shell代码对斑马鱼的每条染色体进行比对,这里贴一条看得更加清楚的代码。
1 | lastz fa/chr1.fa Cyprinus_carpio.fa O=400 E=30 K=3000 L=3000 H=2200 T=1 --format=axt --ambiguous=n --ambiguous=iupac > axt/chr1.axt |
第2步:Chaining
从axt文件得到chain文件
1 | mkdir chain |
上面用了shell代码获得斑马鱼的每条染色体的chain文件,这里贴一条看得更加清楚的代码。
1 | axtChain axt/chr1.axt Danio_rerio.2bit Cyprinus_carpio.2bit chain/chr1.chain -minScore=3000 -linearGap=medium |
合并chain文件,并过滤
1 | chainMergeSort chain/*.chain > all.chain |
第3步:Netting
1 | chainNet all.pre.chain -minSpace=1 Danio_rerio.sizes Cyprinus_carpio.sizes stdout /dev/null | netSyntenic stdin noClass.net |
第4步:Maffing
1 | netToAxt noClass.net all.pre.chain Danio_rerio.2bit Cyprinus_carpio.2bit stdout | axtSort stdin Danio_rerio.Cyprinus_carpio.axt |
总结
经过这4步,就得到了基因组两两比对的maf文件Danio_rerio.Cyprinus_carpio.maf。原教程还有第5步计算Phastcons,不过通常的做法是根据多基因组比对的结果计算Phastcons,所以这部分我们另外讲。我写了一个Python脚本,让大家方便地运行以上步骤。
在docker中使用Dfam TE Tools分析基因组重复序列
手动安装RepeatMasker和RepeatModeler有点手疼,还好Dfam提供了基于docker的TE Tools。
TE Tools的官网:https://github.com/Dfam-consortium/TETools。
使用docker,需要系统管理员将用户添加到docker用户组。
构建镜像
1 | wget https://github.com/Dfam-consortium/TETools/archive/refs/tags/1.4.tar.gz |
构建成功后,可以使用docker image ls查看镜像。
构建镜像考验网速,可以下载我已经构建好的镜像。
1 | wget https://www.biochen.org/public/software/tetools_1.4.tar.gz |
在docker中使用RepeatMasker/RepeatModeler
1 | docker run -it --rm --user $(id -u ${USER}):$(id -g ${USER}) -v /home/chenwen/data:/data dfam/tetools:1.4 |
参数说明
1 | -it 为容器重新分配一个伪输入终端,以交互模式运行容器 |
在启动的新终端中,就可以运行RepeatMasker或者RepeatModeler,数据存放在宿主机的/home/chenwen/data目录中,对应到容器的/data目录。运行完之后,在终端输入exit退出容器。
RepeatMasker的用法参考之前的博文使用RepeatMasker屏蔽基因组重复序列。
RepeatModeler的用法参考之前的博文使用RepeatModeler从头预测基因组重复序列。
使用RepeatModeler从头预测基因组重复序列
简介
官网:http://www.repeatmasker.org/RepeatModeler/
Repeatmasker 基于与已知的重复序列数据库比对来寻找重复序列,Repeatmodeler是通过重续序列的结构特征来进行从头注释,因此可以寻找一些物种特有的重复序列。
RepeatMasker依赖RepBase数据库和Dfam数据库来屏蔽重复序列,对于非模式生物来讲,这两个数据库覆盖有限。通常的做法就是RepeatModeler从头预测重复序列,然后将结果作为RepeatMasker的输入。
安装依赖
RepeatModeler依赖的RepeatMasker、TRF、RMBlast请参考我的上一篇博文使用RepeatMasker屏蔽基因组重复序列,接下来我们只安装剩下的依赖。
RECON - De Novo Repeat Finder
1 | cd ~/software/ |
RepeatScout - De Novo Repeat Finder
1 | cd ~/software/ |
运行LTR结构搜索,还需要安装如下软件。
LtrHarvest - The LtrHarvest program is part of the GenomeTools suite
1 | cd ~/software/ |
CD-HIT - A sequence clustering package
1 | cd ~/software/ |
Ltr_retriever - A LTR discovery post-processing and filtering tool
1 | cd ~/software/ |
LTR_retriever依赖TRF、BLAST+、CD-HIT、HMMER和RepeatMasker。CD-HIT的安装见本文,TRF、HMMER、RepeatMasker的安装见上一篇博文使用RepeatMasker屏蔽基因组重复序列,BLAST+的安装参考另外一篇博文本地BLAST。
编辑~/software/LTR_retriever-2.9.0/目录下的paths文件,写入如下内容,指定这些依赖的安装路径。
1 | BLAST+= /home/chenwen/software/ncbi-blast-2.12.0+/bin # a path that contains makeblastdb, blastn, blastx |
MAFFT - A multiple sequence alignment program.
1 | cd ~/software/ |
编辑~/software/mafft-7.487-with-extensions/core/目录下的Makefile文件,将PREFIX = /usr/local修改为PREFIX = /home/chenwen/software/mafft-7.487,然后继续执行如下命令。
1 | cd ~/software/mafft-7.487-with-extensions/core/ |
Ninja - A tool for large-scale neighbor-joining phylogeny inference and clustering
1 | cd ~/software/ |
UCSC TwoBit Tools
1 | mkdir -p ~/software/kent/ |
安装和配置RepeatModeler
1 | cd ~/software/ |
根据提示配置如下路径
1 | REPEATMASKER_DIR /home/chenwen/software/RepeatMasker |
如果配置成功,将会出现如下提示信息。
1 | Congratulations! RepeatModeler is now ready to use. |
将RepeatModeler添加到环境变量
1 | echo "export PATH=$HOME/software/RepeatModeler-2.0.2a:\$PATH" >> ~/.bashrc |
运行RepeatModeler
这里我们以从Ensembl下载的鲤鱼基因组为例。
1 | mkdir ~/test_RepeatModeler |
建库
1 | cd ~/test_RepeatModeler/ |
参数说明
1 | -name 库的名字 |
运行RepeatModeler
1 | cd ~/test_RepeatModeler/ |
参数说明
1 | -database 库的名字,与上一步一致 |
RepeatModeler的搜索引擎已经只支持RMBlast了,如果使用-engine abblast,RepeatModeler会报出警告WARNING: "-engine abblast" is deprecated, this verison of RepeatModeler uses rmblast only.,然后继续使用RMBlast。
结果解读
我们查看一下日志文件run.out的末尾。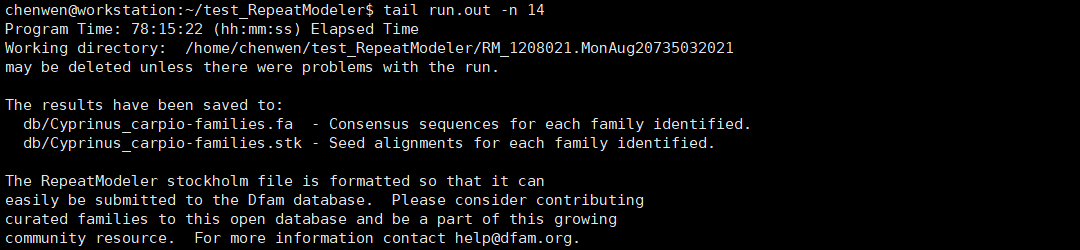
鲤鱼基因组大小约1.7G,RepeatModeler在我的i5 10400(6核12线程)电脑上面跑了约78小时。
RepeatModeler会生成一个名字像这样的文件夹RM_1208021.MonAug20735032021,这是工作目录,保存了中间结果,可以删除。主要结果有两个,db/Cyprinus_carpio-families.fa可以作为library用于RepeatMasker进行重复序列的屏蔽。db/Cyprinus_carpio-families.stk是Dfam兼容的Stockholm格式,可以上传到Dfam数据库。
运行RepeatMasker
我们还需要利用RepeatModeler生成的Cyprinus_carpio-families.fa,使用RepeatMasker屏蔽基因组重复序列。
1 | cd ~/test_RepeatModeler/ |
使用了-lib参数,会使-species参数失效,不需要同时指定-species参数了。使用-lib db/Cyprinus_carpio-families.fa屏蔽了40.02%的重复序列,而使用-species 'Cyprinus carpio'也能屏蔽39.62%的重复序列。二者相差不大,可能是Dfam数据库的重复序列已经覆盖到了鲤鱼。有些非模式生物,单用RepeatMasker只能屏蔽约10%的重复序列,这时使用RepeatModeler就非常有必要了。
使用RepeatMasker屏蔽基因组重复序列
简介
复杂的基因组大部分是可转座元件和其他重复序列,在全基因组比对或者基因注释之前需要屏蔽重复序列,否则就会浪费计算资源,也会得到许多无意义的结果。
RepeatMasker(官网:http://www.repeatmasker.org/RMDownload.html)是一款识别基因组中重复序列和低复杂度序列的软件。RepeatMasker依赖序列搜索引擎(HMMER、Cross_Match、ABBlast或者RMBlast)和重复序列数据库(Dfam和Repbase)来屏蔽重复序列。
对于非模式生物,这两个数据库覆盖有限,因此需要先使用RepeatModeler从头预测重复序列,然后将RepeatModeler预测的序列作为RepeatMasker的输入,来完成最终的重复序列屏蔽。更多的信息请访问RepeatMasker的官网http://www.repeatmasker.org/RepeatModeler/。
安装依赖
本文使用RepeatMasker-4.1.2-p1,它需要以下依赖:
- perl 5.8.0或者更高的版本
- Python 3和h5py库
- 序列搜索引擎
- TRF - Tandem Repeat Finder
- 重复序列数据库
我的HOME路径是/home/chenwen/,软件安装都安装在/home/chenwen/software/,请记得修改为适合自己的路径。
RepeatMasker支持序列4种搜索引擎,我们没有必要全都装上,只安装RMBlast也可以。
安装Cross_Match
按照官网http://www.phrap.org/说明,使用学术机构的邮箱申请软件。申请通过后,软件通过邮件发送给我们。这里提供我的副本phrap_cross_match_swat_1.090518.zip。
1 | cd ~/software/ |
安装RMBlast
官网http://www.repeatmasker.org/RMBlast.html
1 | cd ~/software/ |
安装HMMER
官网http://hmmer.org/,RepeatMasker官方推荐使用HMMER v3.2.1,而不是其他版本。
1 | cd ~/ |
安装ABBlast
在http://blast.advbiocomp.com/licensing/填写表格后,软件和使用许可通过邮件发送给我们。ABBlast解压即可用,使用许可license.xml需要放置到~/.config/ab-blast目录下。
1 | cd ~/software/ |
安装trf
官网https://github.com/Benson-Genomics-Lab/TRF
1 | cd ~/software/ |
安装和配置RepeatMasker
下载RepeatMasker
1 | cd ~/software/ |
下载RepBase数据库
1 | cd ~/software/RepeatMasker/ |
更新Dfam数据库
RepeatMasker带一个小的Dfam数据库,我们需要重新下载完整版的数据库。解压后的Dfam.h5文件83G,非常大。
1 | cd ~/software/RepeatMasker/Libraries/ |
安装h5py
1 | pip install h5py |
配置RepeatMasker
1 | cd ~/software/RepeatMasker/ |
配置过程中,必须配置trf路径,4种搜索引擎的路径可以选择配置。
1 | TRF_PRGM: /home/chenwen/software/trf |
如果配置成功,将有如下提示。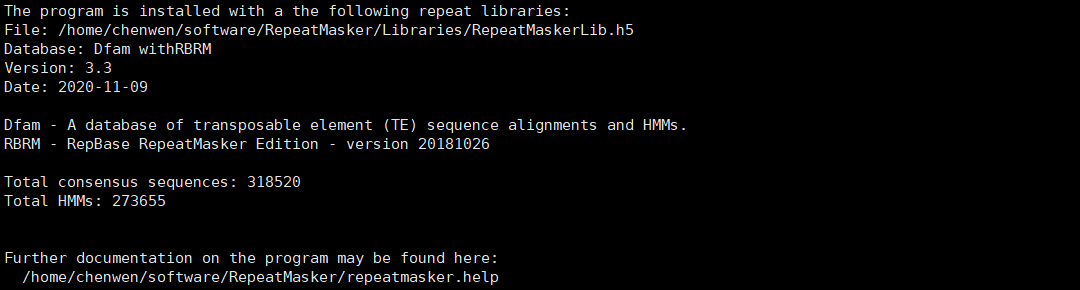
配置成功之后,删掉巨大的Dfam.h5文件。
1 | rm ~/software/RepeatMasker/Libraries/Dfam.h5 |
将RepeatMasker添加到环境变量
1 | echo "export PATH=$HOME/software/RepeatMasker:\$PATH" >> ~/.bashrc |
运行RepeatMasker
这里我们以从Ensembl下载的斑马鱼基因组为例。
1 | mkdir ~/test_RepeatMasker |
1 | RepeatMasker Danio_rerio.fa -species "Danio rerio" -e rmblast -xsmall -s -gff -pa 12 |
参数说明
1 | -e rmblast 指定搜索引擎为rmblast,还可以选择crossmatch、abblast或者hmmer |
结果解读
斑马鱼基因组大小约1.4G,上述命令在我的i5 10400(6核12线程)电脑上面跑了约10.5小时。结果文件主要有:Danio_rerio.fa.tbl、Danio_rerio.fa.masked、Danio_rerio.fa.out、Danio_rerio.fa.out.gff、Danio_rerio.fa.cat.gz。.cat.gz是重复序列与基因组序列的比对文件,其他文件我们打开看一下。
.tbl文件是重复序列的统计信息,我们可以看到斑马鱼有60%以上的重复序列。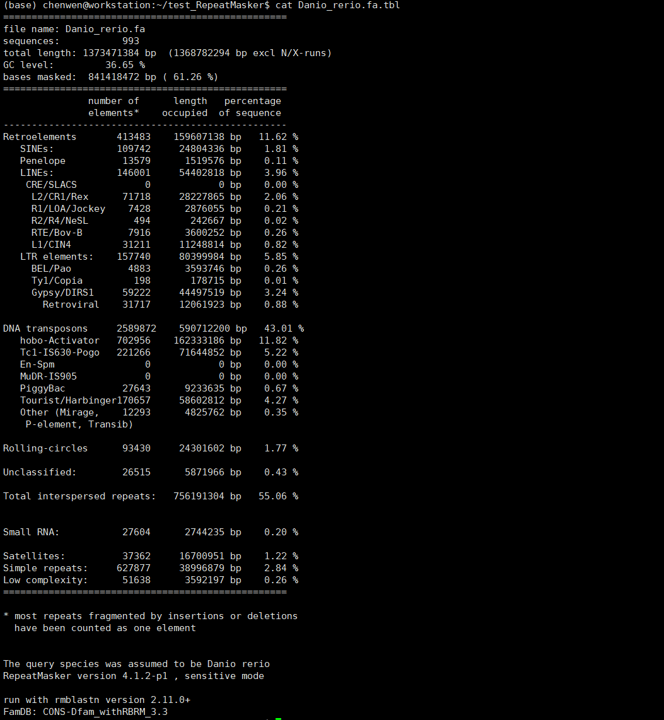
.masked文件是屏蔽重复序列后的基因组文件,这里是软屏蔽,重复序列用小写字母表示。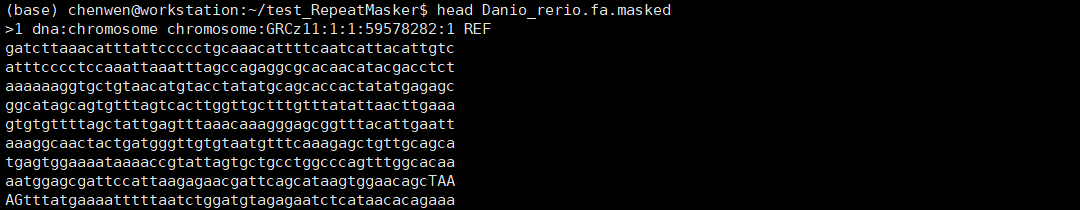
.out文件是RepeatMasker默认的输出结果,.out.gff文件是相应的GFF文件。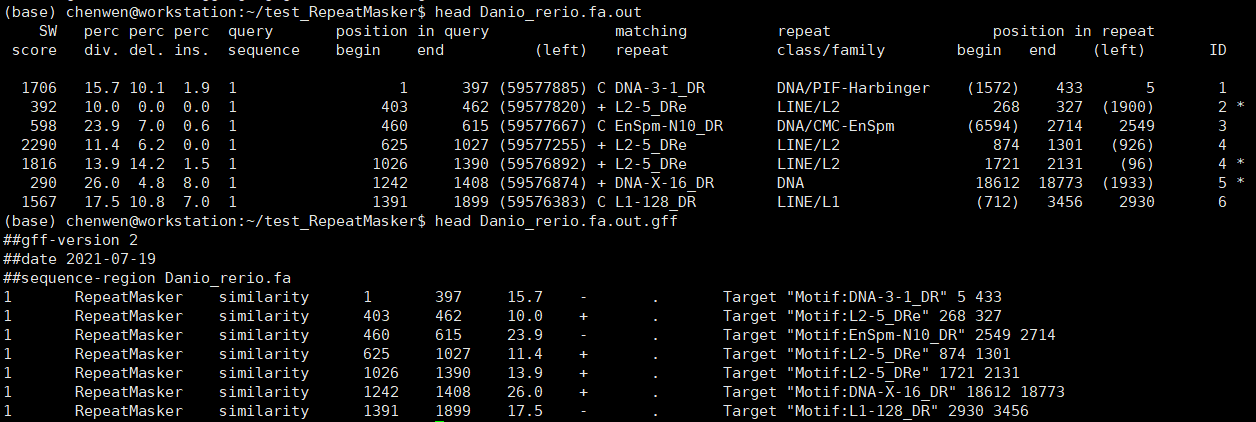
我的主力电脑
可能让大家失望了,我的主力电脑不是服务器平台,也不是HEDT(high end desktop)平台。得益于技术进步,最普通的家用电脑,也能很好的完成我的生物信息分析。
配置单
1 | CPU:英特尔 i5 10400 |
这个配置的价格约6000元,性价比非常高。
使用体验
盒装CPU自带的散热器,全铝鳍片,没有导热铜管,下压式而不是侧吹式,我担心因为散热的问题不能发挥出CPU的全部性能。实际测试,盒装自带的散热器完全够用,i5-10400能跑满全核心睿频4.0GHz,温度在62度左右。
双通道内存有15%左右的性能提升,多线程有12%左右的性能提升。
查看CPU运行频率
1 | sudo apt install cpufrequtils |
查看CPU温度
1 | sudo apt install lm-sensors |
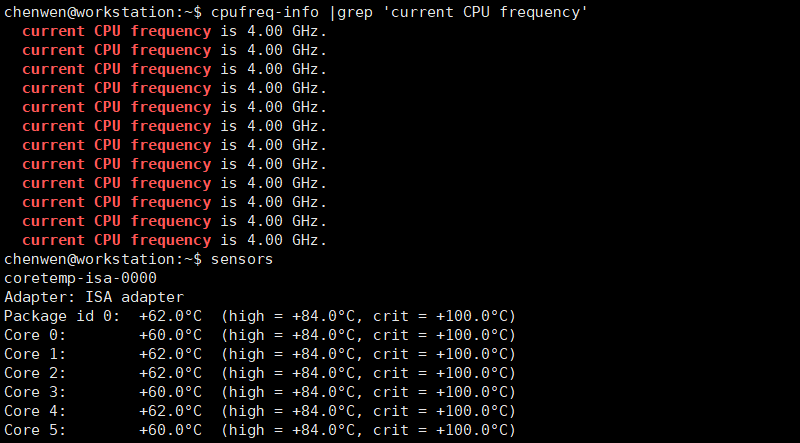
消费级CPU主频高、单核性能好,再配上NVMe固态硬盘,大部分时候的使用体验优于组里的戴尔T640服务器。目前消费级平台最大只支持128G内存,HEDT平台一般可以支持到256G内存,需要更高的内存的话,就需要上服务器平台了。128G内存除了跑大转录组和大基因组的从头组装不行外,其他任务都能胜任。一块4T硬盘不够用,之后又加了一块4T硬盘,以后还需要再加。所有的数据都不能删,硬盘存储是最大的压力。